The directives available are as follows.
directive: meaning:
all pairs use all pairs for electrostatic calculations
cap f cap forces during equilibration period
f is maximum cap in units of kT/Å
(default f=1000)
close time f set job closure time to fseconds
collect include equilibration data in overall statistics
coul calculate coulombic forces
cut f set required forces cutoff to f (Å)
distan calculate coulombic forces using distance dependentdielectric
delr f set Verlet neighbour list shell width to f (Å)
ensemble nve select NVE ensemble (default)
ensemble nvt ber f
select NVT ensemble with Berendsen thermostat
with relaxation constant f (ps)
ensemble nvt evans
select NVT ensemble with Evans thermostat
ensemble nvt hoover f
select NVT ensemble with Hoover-Nose thermostat
with relaxation constant f (ps)
ensemble npt ber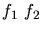
select Berendsen NPT ensemble with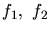
as the thermostat and barostat relaxation times (ps)
ensemble npt hoover
select Hoover NPT ensemble with
as the thermostat and barostat relaxation times (ps)
ensemble nst ber
select Berendsen NTensemble, with
as the thermostat and barostat relaxation times (ps)
ensemble nst hoover
select Hoover N
as the thermostat and barostat relaxation times (ps)
ensemble pmf select (NVE) potential of mean force ensemble
eps f set relative dielectric constant to f(default 1.0)
equil n equilibrate simulation for first ntimesteps
ewald precision f select Ewald sum for electrostatics, with
automatic parameter optimisation ()
ewald sumk1 k2 k3
select Ewald sum for electrostatics, with:Ewald convergence parameter (Å
)
k1
k2
k3
finish close the CONTROL file (last data record)
hke precision f i j select HK-Ewaldsum for electrostatics, with
automatic parameter optimisation (
i
j
hke sum
select HK-Ewald sum for electrostatics, with:
k1
k2
nhko
nlatt
job time f set job time to f seconds
mult n set multiple timestep (multi-step)interval (activated when n2)
no elec ignore coulombic interactions
no vdw ignore short range (non-bonded) interactions
pres f set required system pressure to f k.bars
(target pressure for constant pressure ensembles)
prim f set primary cutoff to f (Å)
(for multiple timestep algorithm only)
print n print system data every n timesteps
print rdf print radial distribution functions
quaternion f set quaternion tolerance to f (default 10)
rdf f calculate radial distribution functions atintervals
of f timesteps
reaction select reaction field electrostatics
restart restart job from end point of previous run
(i.e. continue current simulation)
restart scale restart job from previous run with temperaturescaling
(i.e. begin a new simulation from older run)
scale n rescale atomic velocities every n steps(during equilibration)
shake f set shake tolerance to f (default10
shift calculate electrostatic forces using shiftedcoulombic potential
spme precision f select Ewald sum for electrostatics, with
automatic parameter optimisation (
spme sum
select Ewald sum for electrostatics, with:
k1
k2
k3
stack n set rolling average stack to ntimesteps
stats n accumulate statistics data every ntimesteps
steps n run simulation for n timesteps
temp f set required simulation temperature to f K
traj i j k write HISTORY file with controls:
i
j
k
timestep f set timestep to f ps
zden calculate the z-density profile
zero perform zero temperature MD run