where n is an integer specifying the number of different types of molecule appearing in the FIELD file. Note that enantiomers are counted as two different types of molecule. Once this directive has been encountered, DL_MULTI enters the molecular description environment in which only molecular decription keywords and data are valid.
Immediately following the molecular types directive, are the records defining individual molecules. The first directive is the same as in DL_POLY_2 :
The next directive tells DL_MULTI that there are multipoles in the FIELD file.
The next directive is the same as DL_POLY_2
sitnam a8 atomic site nameThis is the same as the standard DL_POLY_2 . Note however that the atomic site charge read here is ignored as it will be read below in the multipole input. Also the repeat counter will almost always be 1 as atoms in the molecule with the same atomic number will have different multipoles and will be treated by DL_MULTI as different atomic types. ifrz and igrp are not relevant for DL_MULTI use and should be omitted.
weight real atomic site mass
chge real atomic site charge
nrept integer repeat counter
ifrz integer `frozen' atom (if ifrz)
igrp integer neutral/charge group number
The next record carries the entry (without a keyword)
jpoleo i8 multipole pole order for this atom
![]()
jpoleo is the pole order for the multipoles for this atom.
There are then a number of records giving the magnitude of the multipoles depending on jpoleo. The real numbers giving the magnitude of the multipoles are ``f11.0'' fixed format.
In all cases there is a record format ``f11.0'' giving the multipole charge term (order 0).
If
![]() there is a record format ``3f11.0'' giving
the dipole components.
there is a record format ``3f11.0'' giving
the dipole components.
If
![]() there is a record format ``5f11.0'' giving
the quadrupole components.
there is a record format ``5f11.0'' giving
the quadrupole components.
If
![]() there is a record format ``7f11.0'' giving
the octopole components.
there is a record format ``7f11.0'' giving
the octopole components.
If
![]() there are 2 records format ``7f11.0/2f11.0''
giving the hexadecapole components.
there are 2 records format ``7f11.0/2f11.0''
giving the hexadecapole components.
The order of the multipole components follow the usual order for spherical harmonics. [47] Table 1. These are given below.
DL_MULTI does not use the following directives from DL_POLY_2 . bonds, constraints, pmf, angles, dihedrals, inversions teth. For DL_MULTI the records rigid units and mulaxes are described below. Note that the atomic site indices referred to below are indices arising from numbering each atom in the molecule from 1 to the number specified in the atoms directive for this molecule. This same numbering scheme should be used for all descriptions of this molecule. DL_MULTI will itself construct the global indices for all atoms in the systems, as for DL_POLY_2 .
m integer number of sites in rigid unitUp to 15 sites can be specified on the first record. Additional records are used if necessary. Up to 16 sites are specified per record thereafter. The format is 16i5.
site 1 integer first site atomic index
site 2 integer second site atomic index
site 3 integer third site atomic index
.. .. etc.
site m integer m'th site atomic index
mulaxes a40 Define the axis system used to define themultipoles
There is a second keyword on the mulaxes directive.
The mulaxes record must be followed by one record with 9 integer values format 9i5.
direction 1
atom 1a
atom 1b
direction 2
atom 2a
atom 2b
atom 2c
direction 3
enantiomer flag
direction 1, 2 and 3 refer to the x, y and z directions in the
local axis system.
(Use the integer 1 for x, 2 for y and 3 for z, each of the values
must be used once).
Usually direction 1 will be 1, direction 2 will be 2 and direction
3 will be 3, and this must be the order if the enantiomer flag
is 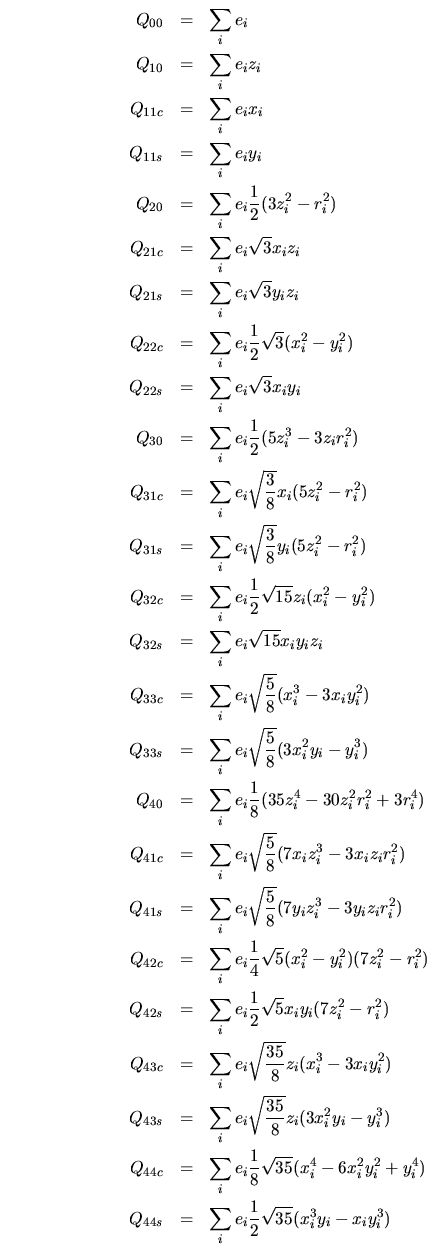 .
The axis in direction 1 (normally x axis) is defined along
the bond from atom 1a to atom 1b.
The axis in direction 2 (normally y axis) is defined in the
plane containing atoms 2a, 2b and 2c, and normal to the axis
in direction 1.
The axis in direction 3 ( normally z axis) makes a right handed
set with 1 and 2.
The enantiomer flag can have values 1 or -1.
.
The axis in direction 1 (normally x axis) is defined along
the bond from atom 1a to atom 1b.
The axis in direction 2 (normally y axis) is defined in the
plane containing atoms 2a, 2b and 2c, and normal to the axis
in direction 1.
The axis in direction 3 ( normally z axis) makes a right handed
set with 1 and 2.
The enantiomer flag can have values 1 or -1.
In many crystalline systems the crystal structure contains a mixture of two enantiomers (at least experimentally). Often this will be the result of a slight departure from planarity in the experimental results which may or may not be physically real. To permit the user the option of inputting a pair of enantiomers, they are treated by DL_MULTI as two separate molecules. However, the setting up of the molecular axes using molaxes will set up two right handed axes systems which are not mirror images of each other. To allow for this the signs of the appropriate multipole components need to be changed. There are two ways the user can do this. First, the user can change the sign of all the odd-z components in the multipole description of the second molecule. Secondly, the user can use the enantiomer flag -1 and leave the components as they are. DL_MULTI will then change the sign of the odd-z components. Note that if a non-standard order of the axes in molaxes is used, then the first method must be used. The user will need to change the sign of the odd-x or odd-y components for the second molecule (according to which axis is defined third in the description).
The entries for a second molecule may now be entered, beginning with the name-of-molecule record and ending with the finish directive.
The cycle is repeated until all the types of molecules indicated by the molecules directive have been entered.
The user is recommended to look at the example FIELD files in the data directory to see how typical FIELD files are constructed.