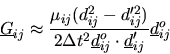![]()
![]()
The SHAKE algorithm for bond constraints was devised by Ryckaert et al. [13] and is widely used in molecular simulation. It is
a two stage algorithm based on the Verlet integration scheme [12]. DL_POLY_2 employs a leapfrog variant. In the first
stage the Verlet leapfrog algorithm calculates the motion of the atoms
in the system assuming a complete absence of the rigid bond forces. The
positions of the atoms at the end of this stage do not conserve the distance constraint
required by the rigid bond and a correction is necessary. In the second
stage the deviation in the length of a given rigid bond is used
retrospectively to compute the constraint force needed to conserve the bondlength. It is
relatively simple to show that the constraint force has the form
where: ![]() is the reduced mass of the two atoms connected by the bond;
is the reduced mass of the two atoms connected by the bond; ![]() and
and ![]() are the original and
intermediate bond vectors;
are the original and
intermediate bond vectors; ![]() is the constrained bondlength; and
is the constrained bondlength; and ![]() is the Verlet integration time step. It should be noted that this formula is an
approximation only.
is the Verlet integration time step. It should be noted that this formula is an
approximation only.
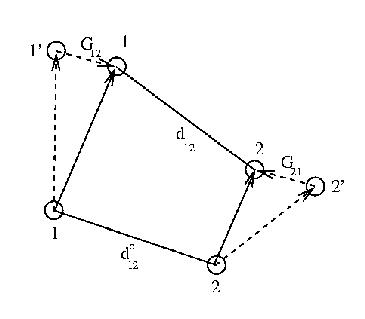
The SHAKE algorithm calculates the constraint force ![]() that conserves the
bondlength
that conserves the
bondlength ![]() between atoms
between atoms ![]() and
and ![]() , following the initial movement to positions
, following the initial movement to positions ![]() and
and ![]() under the unconstrained
forces
under the unconstrained
forces ![]() and
and ![]() .
.
For a system of simple diatomic molecules, computation of the constraint force will, in
principle, allow the correct atomic positions to be calculated in one pass. However in the
general polyatomic case this correction is merely an interim adjustment, not only because
the above formula is approximate, but the successive correction of other bonds in a
molecule has the effect of perturbing previously corrected bonds. The SHAKE algorithm is
therfore iterative, with the correction cycle being repeated for all bonds until each has
converged to the correct length, within a given tolerance. The tolerance may be of the
order ![]() to
to ![]() depending on the precision desired.
depending on the precision desired.
The procedure may be summarised as follows:
The parallel version of this algorithm, as implemented in DL_POLY_2 , is known as RD_SHAKE [11] (see section 2.6.8). The subroutine NVE_1 implements the Verlet leapfrog algorithm with bond constraints. The routine RDSHAKE_1 is called to apply the SHAKE corrections to position.
It should be noted that the fully converged constraint forces ![]() make a contribution
to the system virial and the stress tensor.
make a contribution
to the system virial and the stress tensor.
The contribution to be added to the atomic virial (for each constrained bond) is
| (2.179) |
The contribution to be added to the atomic stress tensor is given by
| (2.180) |
where ![]() and
and ![]() indicate the
indicate the ![]() components. The atomic stress tensor derived from the pair
forces is symmetric.
components. The atomic stress tensor derived from the pair
forces is symmetric.
![]()