![]()
![]()
For simulations employing a large spherical cutoff ![]() in the calculation of the
interactions DL_POLY_2 offers the possibility of using a multiple timestep algorithm to
improve the efficiency. The method is based on that described by Streett et al [39,40] with
extension to Coulombic systems by Forester et al [41].
in the calculation of the
interactions DL_POLY_2 offers the possibility of using a multiple timestep algorithm to
improve the efficiency. The method is based on that described by Streett et al [39,40] with
extension to Coulombic systems by Forester et al [41].
In themultiple timestep algorithm there are two cutoffs for the pair
interactions: a relatively large cutoff (![]() ) which is used to define the standard Verlet
neighbour list; and a smaller cutoff
) which is used to define the standard Verlet
neighbour list; and a smaller cutoff ![]() which is used to define a primary
list within the larger cutoff sphere (see figure). Forces derived from atoms in the
primary list are generally much larger than those derived from remaining (so-called secondary)
atoms in the neighbour list. Good energy conservation is therefore possible if the forces
derived from the primary atoms are calculated every timstep, while those from the
secondary atoms are calculated much less frequently, and are merely extrapolated over the
interval. DL_POLY_2 handles this procedure as follows.
which is used to define a primary
list within the larger cutoff sphere (see figure). Forces derived from atoms in the
primary list are generally much larger than those derived from remaining (so-called secondary)
atoms in the neighbour list. Good energy conservation is therefore possible if the forces
derived from the primary atoms are calculated every timstep, while those from the
secondary atoms are calculated much less frequently, and are merely extrapolated over the
interval. DL_POLY_2 handles this procedure as follows.
DL_POLY_2 updates the Verlet neighbour list at irregular intervals,
determined by the movement of atoms in the neighbour list (see section 2.1). The interval between updates is usually of the
order of ![]() 20 timesteps. Partitioning the Verlet list into primary
and secondary atoms always occurs when the Verlet list is updated, and thereafter at
intervals of multt timesteps (i.e. the multi-step interval specified by the user
- see section 4.1.1). Immediately after the
partitioning, the force contributions from both the primary and secondary atoms are
calculated. The forces are again calculated in total in the subsequent timestep. Thereafter, for multt-2 timesteps, the forces derived from the
primary atoms are calculated explicitly, while those derived from the secondary atoms are
calculated by linear extrapolation of the exact forces obtained in the first two timesteps
of the multi-step interval. It is readily apparent how this scheme can lead to a
significant saving in execution time.
20 timesteps. Partitioning the Verlet list into primary
and secondary atoms always occurs when the Verlet list is updated, and thereafter at
intervals of multt timesteps (i.e. the multi-step interval specified by the user
- see section 4.1.1). Immediately after the
partitioning, the force contributions from both the primary and secondary atoms are
calculated. The forces are again calculated in total in the subsequent timestep. Thereafter, for multt-2 timesteps, the forces derived from the
primary atoms are calculated explicitly, while those derived from the secondary atoms are
calculated by linear extrapolation of the exact forces obtained in the first two timesteps
of the multi-step interval. It is readily apparent how this scheme can lead to a
significant saving in execution time.
Extension of this basic idea to simulations using the Ewald sum requires the following:
In this way the Coulombic forces can be handled by the same multiple timestep scheme as the van der Waals forces. The algorithm is described in detail in [41].
Note that the accuracy of the algorithm is a function of the multi-step interval multt, and decreases as multt increases. Also, the algorithm is not time reversible and is therefore susceptible to energy drift. Its use with a thermostat is therefore advised.
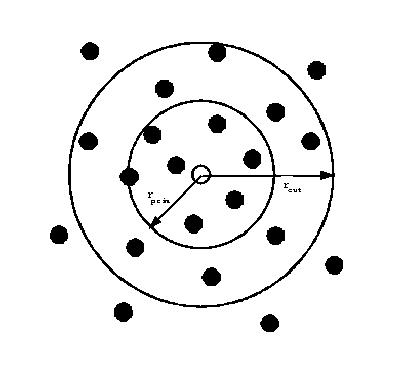
The multiple timestep algorithm
The atoms surrounding the central atom (open circle) are classified as primary if they
occur within a radius ![]() and secondary if outside this radius but within
and secondary if outside this radius but within ![]() .
Interactions arising from primary atoms are evaluated every timestep.
Interactions from secondary atoms are calculated exactly for the first two steps of a
multi-step and by extrapolation afterwards.
.
Interactions arising from primary atoms are evaluated every timestep.
Interactions from secondary atoms are calculated exactly for the first two steps of a
multi-step and by extrapolation afterwards.
![]()